
Simulations to Introduce Protein Flexibility
Nupur Bansal is currently a fifth-year Ph.D. student pursuing research in computational Chemistry. For the past five years she has been working under Dr. Kenneth M. Merz Jr., studying protein flexibility in the binding pocket region. Her research interest is accurately predicting protein ligand binding free energies by introducing conformational sampling in the protein’s binding pocket region. Bansal refers to the accurate prediction of binding free energy as “the holy grail of the computer-aided drug design field.”

Her work will be especially beneficial for proteins like Kinase and HIV, which have huge loops (unstructured segments in proteins that are most mobile). Pharmaceutical industries are trying to target the loops in these proteins to design better drugs and inhibitors. As these loops are quite mobile, it is quite difficult to recognize their role in designing an inhibitor. “The hope is that once we can estimate the energetic contribution of loop in protein-ligand binding mechanism, it would be extremely helpful to pharmaceutical industries” states Bansal.
In general, proteins have huge degrees of freedom, due to their excessive number of atoms. This makes it computationally very expensive to track all the motions that they can undergo. Bansal has developed a tool that can introduce flexibility of proteins in the binding pocket region. She wrote her code using MATLAB software, and ran it on HPCC.
Bansal has performed two separate applications regarding protein loop flexibility. The first application includes the side chain receptor flexibility on a set of 159 protein-ligand complexes. Bansal says incorporating the receptor flexibility resulted in, “improvements in both structural RMSDs (Root mean squared deviations) of docked ligand and predicted binding free energy calculations.” Structural RMSD is a way to access the quantitative difference between the two given structures. “In our case, it was calculated between the ligand from experimental co-crystal structure and the docked ligand and it improved after receptor flexibility was incorporated as compared to the case when receptor flexibility was not included,” states Bansal.
The second application focused on the effect of loop motions on the streptavidin-biotin system. Including both side chain and backbone flexibility on the eight residue long loop in Streptavidin, Nupur was able to study the effect of loop on the binding of biotin to Streptavidin. It was here that she came across the challenge of open and closed loops. The structural difference between the two is huge, making it computationally very difficult to track all the motions. In the second application, backbone flexibility was incorporated which made the conformation generation quite slow because of backbone connectivity and added degrees of freedom. However, using her method, she and her colleagues generated the loop conformational ensemble covering both the open and closed states. Conformational ensemble refers to the generated collection of structures. To break it down, there is one eight residue long loop (the unstructured segment) in Streptavidin, which exists in open conformation in Biotin (ligand) unbound state. However, as soon as Biotin comes and binds, the loop closes like a lid. “Imagine this mechanism like the lid of the box getting opened and closed. We generated the loop conformational ensemble which covered all the snapshots of the loop in going from open to closed transition.” states Bansal.
Bansal’s prior work was on molecular dynamic simulations, which was strictly application-based, while her current research on proteins flexibility is application- and development-based. This required her to write her own program. It was her first time writing code, and she claims that her research could not have been completed without assistance from the ICER and HPCC staff and their resources.
Nupur Bansal has a passion for her research, and plans to continue with it in the future. She has a postdoctoral offer from Biogen, an American Biotechnology company. She will be applying her Ph.D. knowledge there by performing computational studies of biological systems.
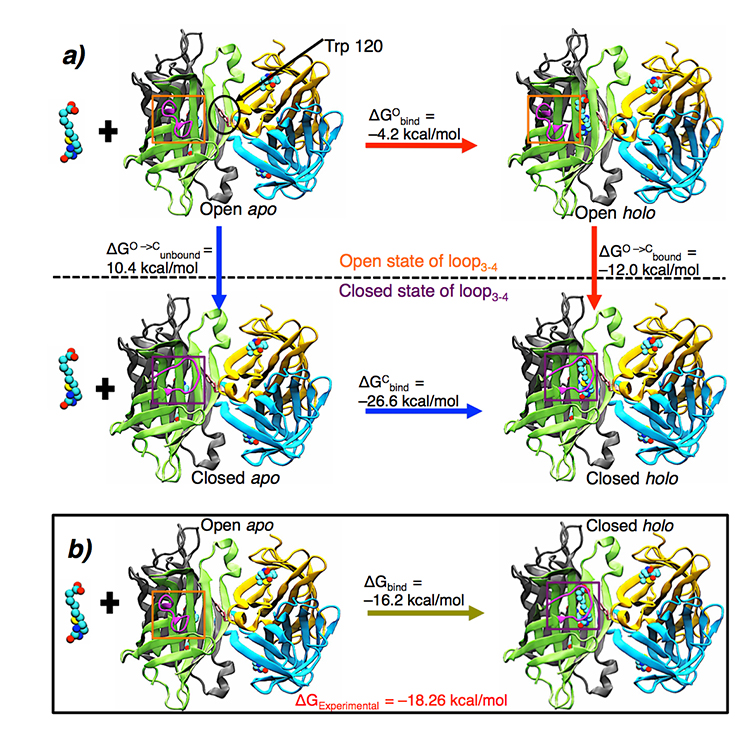
a) Detailed representation of thermodynamic free energy cycle for binding in solution phase for the streptavidin-biotin system.
b) The net free energy change upon biotin binding and loop closure in the solution phase.